
随着 2025 年诺贝尔化学奖对网状化学领域的认可,金属有机框架(MOFs)与共价有机框架(COFs)的研究再次成为材料科学的焦点。
共价有机框架(Covalent Organic Frameworks, COFs)是一类由轻元素通过强共价键连接形成的晶态多孔材料,自 2005 年被首次报道以来,因其多孔性、低密度、高比表面积、结构可调和化学稳定性,在气体分离、催化、储能、光电子学及药物递送等领域展现出巨大潜力。然而,与金属有机框架(MOFs)相比,COFs的研究进展相对缓慢,实验报道的 COFs 仅约 1, 250 种,而 MOFs 已超过 125, 000 种。这一差距的背后,既有合成结晶度的挑战,也有数据积累和计算工具滞后的因素。
这种数据量的匮乏导致早期 AI 研究主要集中在 MOFs 上。然而,随着假设 COF(hypoCOF)数据库的建立(如 Berkeley COF, ReDDCOFFEE)以及高通量计算筛选(HTCS)的发展,AI 正在填补这一空白。虽然 MOFs 在 AI 辅助研究中起步更早、数据库更庞大,但 COFs 凭借其无金属组成、高化学稳定性和模块化设计的独特优势,正迎来 AI 驱动的黄金时代。
本次分享,我们深度解读发表于 JACS Au 的前瞻性综述 《Digital Reticular Chemistry: How Artificial Intelligence Is Redefining COF Research》,梳理 AI 如何从性能筛选、合成智能到自主发现,全方位加速 COFs 材料的设计与应用,为关注多孔材料交叉研究的读者提供参考。


01 从大规模筛选到数据高效的机器学习
在 MOF 领域,高通量计算筛选结合机器学习已发展得较为成熟。研究者建立了多个公开数据库,并利用巨正则蒙特卡洛(GCMC)或分子动力学模拟生成训练数据。COF 领域虽然起步较晚,但也陆续构建了若干假设结构数据库,例如 Berkeley COF 数据库(约 7 万种)、ReDDCOFFEE(约 27 万种)和 Genomic COF(约 47 万种)。这些数据库为机器学习提供了必要的样本空间。

图1 金属有机框架(MOF)和共价有机框架(COF)研究从网状化学到数字化学的演变:网状化学传统上依赖于实验和人类直觉,而数字化学现在则利用大数据集、高通量分子模拟和机器学习模型,以实现材料的快速预测、筛选和逆向设计
早期工作多采用基于描述符的模型,即人为定义孔径、表面积、元素组成等特征,再与模拟或实验数据建立映射。这类方法在面对新任务时往往需要重新训练,且依赖高质量的描述符设计。近来,研究者开始探索更高效或更通用的策略:
• 主动学习:
在 CH₄ 存储筛选中,主动学习算法从 Genomic COF 的 44 万余种结构中仅通过约 50 次 GCMC 模拟就识别出性能最优的材料,其甲烷可交付容量(222.2 v/v)超过了此前已知的最佳 Berkeley COF(216.9 v/v)。这种迭代采样策略显著降低了计算成本。
• 迁移学习:
针对 COF 数据不足的问题,研究者利用在 190 万种多孔材料上预训练的 Transformer 模型,再微调用于 COF 的 H₂ 吸附预测。结果表明,从 MOF 学习到的表征可以迁移至 COF,在数据有限时仍能保持较好的预测精度。
• 直接结构学习:
部分工作不再依赖人工描述符,而是将 COF 的晶体结构文件(CIF)或势能面体素网格直接输入神经网络。例如,使用卷积神经网络处理三维势能网格,在预测 CH₄ 吸附时,仅用较少训练样本即达到甚至优于传统随机森林模型的精度。另一种方法将原子坐标和元素种类视为点云,利用深度学习直接从原始结构预测吸附量,同样在 COF 案例中表现出良好性能。
不过需要指出,这些模型输入的 CIF 通常代表理想化的周期性结构,未考虑实际二维 COF 中层间堆积无序或偏移的影响,这可能高估或低估实际的吸附性能。
02 AI赋能的多场景应用突破
• 气体分离与存储
ML 模型已成功应用于 COFs 的 CH₄、H₂、CO₂ 吸附性能预测。主动学习策略使得仅需模拟 50 种材料即可从近 45 万种候选物中筛选出最优材料;迁移学习则实现了从 MOFs 到 COFs 的知识迁移,缓解了 COF 数据稀缺问题;而基于晶体结构直接输入的深度学习方法(如 3D 体素化、点云表示),进一步摆脱了对人工设计描述符的依赖。

图2 用于COFs中气体分离和气体储存的机器学习方法:a)基于描述符的机器学习模型用于模拟COF膜;b)主动学习用于预测COFs中CH4的传递;c)迁移学习用于评估COFs的氢气(H2)吸收能力;d)直接结构学习用于计算COFs对CH4的吸收
• 催化、光电子学与能源领域
AI在COF研究中另一个活跃的领域是催化、光电子学和能源相关应用,数据往往来自密度泛函理论(DFT)计算,成本较高,因此数据高效的机器学习显得尤为重要。一个典型案例是 H₂O₂ 光合作用 COF 光催化剂的发现。研究者首先基于 DFT 计算获得分子描述符,训练模型后对约 3.3 万种 COF 进行筛选,随后合成了排名靠前的候选材料。其中性能最佳的 TAPPy-COF 在可见光下的 H₂O₂ 产率达到 7.0 mmol g⁻¹ h⁻¹,优于已有报道的 COF 光催化剂,且结构稳定。
在荧光 COF 的发现中,主动学习被用于直接指导实验:贝叶斯优化算法在仅有少量初始数据的情况下,从 3 种三醛与 26 种二胺的组合空间中推荐了 15 种 COF 进行合成,结果发现两种材料的荧光量子产率分别达到约 55% 和 45%,超越了此前的最好水平。这表明即使在化学空间相对较小、初始数据极少时,机器学习仍能有效识别异常高性能材料。

图3 人工智能辅助的COF设计与发现,用于催化、光电子和能源应用:a)基于机器学习的光催化制备H202催化剂设计;b)利用人工智能辅助发现高荧光COFs;c)基于机器学习的串联吸附式热泵用分子筛膜MOF和COF对筛选
• 环境与生物医学应用
在环境领域,短链 PFAS(如全氟丁酸)的水处理是一个难题。研究者用蒙特卡洛模拟计算了部分 COF 的吸附选择性,再训练晶体图卷积神经网络预测剩余结构,发现三嗪基 COF 对 PFBA 选择性最高,而硼基 COF 虽然选择性也不错但水稳定性差。这类分析不仅提供了排序,还指出了值得优先研究的化学类型。

图4 基于人工智能的COF筛选在环境和生物医学中的应用:a)利用机器学习加速的高通量筛选,用于从水中去除短链全氟和多氟化合物(PFAS);b)深度神经网络引导的COF筛选,用于5-氟尿嘧啶(5-FU)负载
在生物医学方面,针对 5-氟尿嘧啶药物负载的预测,深度神经网络表明比表面积和孔径是主要控制因素,并筛选出 COF-362 作为吸附焓最高的候选。不过作者也提到,药物释放过程通常需要分子动力学模拟,计算成本较高,未来若能结合加速 MD 的 AI 方法,将更有助于实际应用。
03 合成智能:
从条件优化到自主发现
近年来,COF 研究的一个显著趋势是将 AI 从单纯的性能预测拓展到合成条件的优化与设计。传统合成 COF 需要精确控制溶剂、催化剂、温度、时间和化学计量比,这些参数通常依赖文献经验或反复试验。
较早的工作聚焦于已知 COF 的条件优化。例如,在用于大气集水 COF-323 的合成中,研究者利用基于 ChatGPT 的助手进行文献检索和实验规划,配合贝叶斯优化推荐微波合成条件。经过 82 次实验,AI 引导的合成方案使 BET 表面积达到 926–1459 m²/g,约为此前报道最高值的两倍,且在 10–40% 相对湿度下的水工作容量显著提升。
更大规模的尝试是将文献中的合成知识系统化。研究者从 800 多篇 COF 论文中提取了 2709 个合成方案,包括连接体组合、溶剂、催化剂、温度、时间及 PXRD 结果等,构建了一个可检索的数据库。采用检索增强生成(RAG)的方法,对新的连接体对推荐合成条件。基准测试显示,推荐溶剂和催化剂与文献成功条件的一致性约为 83%,实验验证也成功合成了两种新型氟化 COF。
更进一步,LLM加速合成技术(LFAST) 将大语言模型与机器人合成及高通量 PXRD 分析整合。面对一种被 118 篇论文报道但始终未获得尖锐衍射峰的 COF(TpPa-SO₃H),该系统实现了迄今最高的结晶度指数。随后,系统在没有文献先例的情况下,提出并成功合成了一种全新 COF——COF-2000,展示了从推荐条件到自主发现的跨越。
另一项工作中,AI 被用于为特定性能目标选择构筑基元。基于对 355 篇 COF 光催化论文的文本挖掘(提取超过 1.1 万个化学关系),系统推荐了 TAPT 和 BTT 作为构筑单元,并合成了亚胺连接的 Imi-COF 和噻唑连接的 Thz-COF,后者在 H₂O₂ 产率和结构稳定性上均更优。
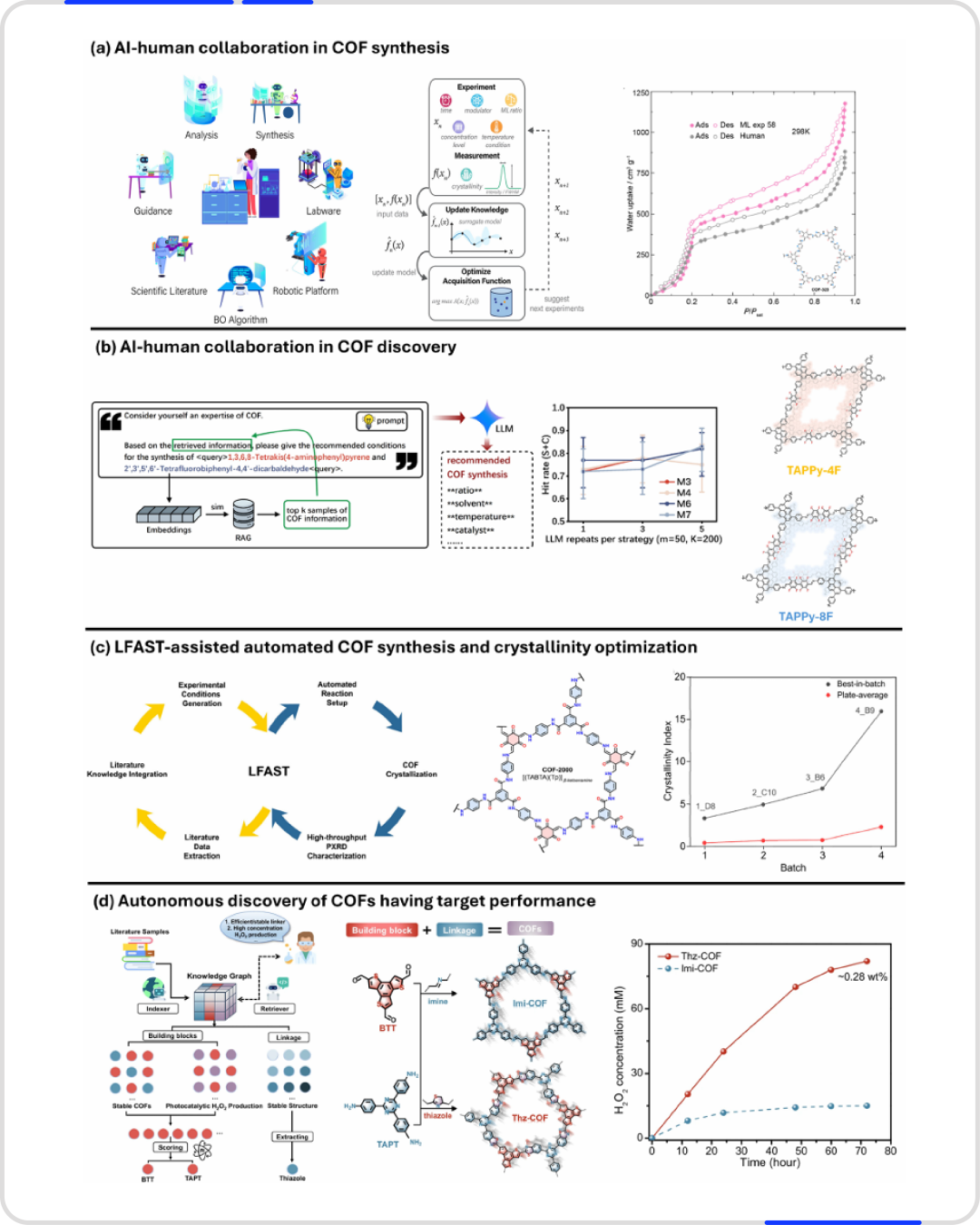
图5 人工智能辅助的人工引导型COF合成、发现与优化:a)人工智能与人工协作,对COF-323的合成条件进行迭代优化,以提高其结晶度和水吸附能力;b)人工智能与人工协作,用于合成新型氟化COFs;c)LFAST辅助的自动化COF合成与结晶度优化;d)具有光催化制氢目标性能的COFs的自主发现
不过作者也谨慎指出,仍需保持清醒。当前的 AI 模型大多基于完美的晶体结构(CIF 文件),当前大语言模型并未真正理解 COF 的成核与生长机制,其推荐主要基于文献中的统计关联,而非物理或化学机理。现实中的 COFs 往往存在层间堆积无序、缺陷和稳定性问题。此外,缺乏 “失败实验” 的数据,也让 AI 对合成难度的评估存在偏差。
因此,AI 预测与文献结果的高度一致性并不等同于真正的 “发现”,严格的实验验证和跨体系的基准测试仍然是必需的。未来的关键,在于建立包含失败案例的标准化数据库,开发能理解复杂拓扑结构的生成式 AI,并最终实现计算与实验的闭环。
04 挑战与未来方向
尽管上述进展令人鼓舞,该展望也明确指出了若干瓶颈:
• 数据质量与偏差:
失败的合成、低结晶度或无定形产物很少被报道,导致训练数据偏向成功案例,模型可能高估合成可行性。未来的数据库应系统收录负面结果,并增加标准化的合成与表征标签。
• 结构表示与拓扑泛化:
大多数模型假设理想晶体结构,对二维 COF 的层间堆叠无序、柔性或缺陷缺乏有效描述。此外,常见拓扑结构(如 hcb、sod)容易被编码,但较复杂的网络(如 qom、ana)则难以推广。改进拓扑感知表示将是提升生成模型可靠性的关键。
• 计算与实验的闭环:
作者强调,未来的自主实验室应实现 “设计—合成—表征—再设计” 的闭环,这需要计算科学家与实验化学家的深度协作。同时,文本挖掘工具(如针对 COF 的 ChemicalTagger 或 MOF-ChemUnity)应扩展到捕捉稳定性限制、活化失败等信息,以训练更具 “合成意识” 的 AI 模型。
• 机器学习势的发展:
目前针对 COF 的机器学习势函数仍很有限,而 COF 的共价键特性、层间弱相互作用及结构柔性都需要专用势能模型。结合量子力学计算、原子模拟和机器学习的多尺度方法有望更真实地预测吸附、扩散及结构响应。
总体来看,AI 在 COF 领域的角色正从加速计算筛选扩展到辅助实验设计和合成规划。但它的作用仍是辅助性的 —— 将研究者的精力从重复性试错中解放出来,而非取代对化学本质的理解。对所有材料科学的前沿探索者来说,拥抱 “数字网状化学”,意味着我们正站在下一个重大科学突破的门槛上。
文献原文:
https://doi.org/10.1021/jacsau.6c00576
声明:
本文所有内容旨在学术探讨与信息交流,所有权利归原作者所有。
从学术前瞻到产业落地,
晶泰科技的AI+材料研发布局
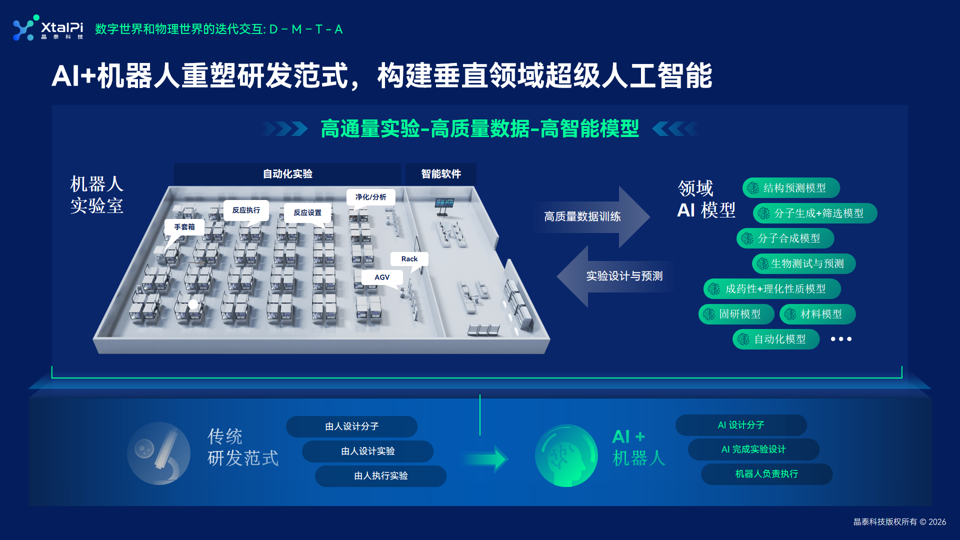
学术界的展望为我们勾勒了 AI 辅助 COF 研究的诸多可能性,但一个不容忽视的现实是:这些方法大多仍停留在计算筛选和有限实验验证阶段。从 AI 推荐到实验室可靠合成,中间还横亘着数据碎片化、实验通量不足、反馈周期长等障碍。要真正跑通 “预测—实验—迭代” 的闭环,不仅需要更聪明的算法,也需要标准化的实验数据生成能力和自动化的执行平台。晶泰科技的算法体系以 AI 为核心,构建了 “干湿闭环” 的科研体系,在药物、材料等多领域实现技术落地。
1. 核心算法布局:200+垂类AI化学模型打造“化学超级大脑”
依托百万级化合物与反应数据库,打造覆盖分子生成、结构优化、反应预测、性质预判、条件筛选等全研发场景的 200 + 垂类 AI 化学模型,融合量子力学计算与机器学习能力,自由能微扰计算精度行业领先,可精准预判反应可行性、分子成药性与材料理化性质,还能自主遍历海量化学空间生成最优实验方案,实时复盘数据修正参数。

2. 算法落地核心:“干湿闭环”实现AI与实验双向赋能
• 干实验(AI 算力)指导湿实验:
AI 提前预判最优反应路径、筛选核心参数,让自动化实验精准靶向有效研发方向
• 湿实验反哺干实验:
自主实验室产生的海量结构化真实数据持续训练、迭代 AI 模型,实现越用越准的自进化效果,打通 “AI 预测 - 智能设计 - 自动化实操 - 数据迭代” 的闭环
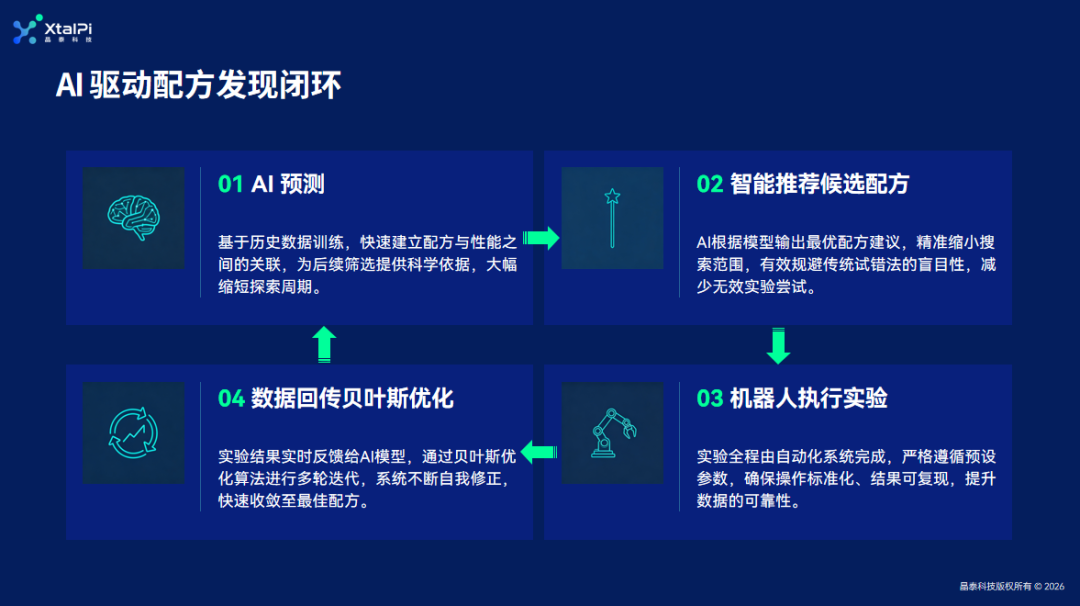
3. 材料领域算法应用案例
• 高分子材料配方优化:
基于客户历史数据建立 AI 预测模型并推荐配方,硬件实现全流程自动化,通过表征结果自主推荐下一轮实验,仅需 3-6 次迭代即可锁定最佳配方,实现 AI + 高分子自动化合成的数据闭环
• MOF 材料研发:
将结构表征与性能测试直接关联,构建以性能为导向的数据闭环,实现 MOF 材料从随机试错向理性设计与快速迭代的跨越
• 催化材料研发:
搭建全流程自动化高通量研发平台,通过 XRD 样品自动制备与表征,系统自动比对分析谱图、判断实验是否达标,结合评价结果利用算法挖掘分子筛结构与催化性能的构效关系
• 电解液配方优化:
先利用通用 AI 模型提取并结构化公开文献数据,再通过图神经网络构建高精度性质预测模型并智能采样,最后由自动化工站执行方案并通过主动学习策略实时优化模型精度

图 AI+机器人结合赋能材料科学研究
4. 算法数据支撑:AI4Science 数据基座与标准化数据基建
• 生物材料研发自主实验室通过端到端自动化、复杂操作标准化和 AI4Science 数据基座,打通 “自动化实验→数据沉淀→模型迭代→智能决策” 的 AI 研发闭环
• 自主研发为 AI 算法而生的数据基础设施,将材料结构、配方、工艺等关键参数编码化,构建专属结构化实验数据库,解决传统实验数据碎片化、非结构化的难题
上一篇:没有了
 联系人:晶泰科技
联系人:晶泰科技 邮箱:bd@xtalpi.com
邮箱:bd@xtalpi.com 电话:021-68780786
电话:021-68780786 地址:深圳市福田区福保街道福保社区红柳道2号国际生物医药产业园二期1层
地址:深圳市福田区福保街道福保社区红柳道2号国际生物医药产业园二期1层
晶泰科技服务号

晶泰智造公众号
©2026 深圳晶泰科技有限公司 版权所有 备案号:粤ICP备17120953号 技术支持:化工仪器网 Sitemap.xml 总访问量:115995 管理登陆
